

Introduction
In order to understand the mechanisms involved in the regulation of hair follicle development, growth, differentiation, and cycling, it is important to have available as many model systems as possible, including in vivo animal models, cell and organ culture.
Each of the above model systems has its advantages and disadvantages. In vivo models allow follicles to be studied in situ and detailed observations can be made using morphological criteria, immunohistochemistry, in situ hybridisation, and RT-PCR. However, it is difficult to quantify molecular or biochemical parameters of individual follicles and in vivo models do not allow follicles to be studied in a fully defined environment. Cell culture involves the isolation and culture of individual cell populations from which many morphological, biochemical, and molecular parameters can be measured. However, cell culture results in the loss of the normal three-dimensional architecture of the organ and as such, it is sometimes difficult to translate observations from cell culture to the whole organ. Histiotypic culture, in which different cell types may be co-cultured, goes some way towards addressing some of the disadvantages of cell culture but still do not fully mimic the in vivo environment.
What Is Organ Culture?
Organ culture involves the in vitro culture of either an intact organ in the case of embryonic tissues or, in the case of neonatal or adult tissue, usually small pieces of an organ.1 The advantage of organ culture is that the component cells of the organ are maintained in their in vivo three-dimensional environment and that often the external environment in which the organ is maintained is fully defined. However, disadvantages include lack of blood supply, associated systemic factors, and innervation. Perhaps the most obvious of these is blood supply. For an organ-maintained tissue to survive, gases, nutrients, growth regulatory factors, and waste products must be able to freely diffuse through the tissue. Therefore, if the organ to be maintained is too large, then the cells in its centre will not be sufficiently perfused, and as a result they will become necrotic and die, resulting in loss of integrity of the cultured organ. Because of these size restrictions, organ culture is frequently used for embryonic tissues and less so for adult organs. However, adult skin, its glands, and appendages, and especially the hair follicle, are an ideal size for organ culture.
Background to Hair Follicle Culture
In 1922, Drew described the in organ maintenance of skin on blood clots. This paper pioneered the organ culture of skin and over the following years a number of researchers used organ cultured skin to investigate many aspects of skin biology including studies of hair follicle development.2,3 These and later studies showed that when embryonic skin was maintained in vitro, hair follicles continued to develop.4-12 However, the ability of cultured embryonic skin to sustain complete hair follicle development generally decreased, both with time in culture and with increasing age of the embryo from which the skin was removed and, moreover, subsequent hair fibre production was rarely observed.
The in vitro growth of hair follicles in post-embryonic skin has also been studied although not to the same extent as embryonic skin.13-15 However, as with embryonic skin, hair follicles in cultured neonatal skin also grew little hair in vitro, when compared with normal in vivo rates. Morphological studies carried out by Frater on skin plugs from rats aged 12 to 14 days of age, did however, show that follicles maintained in vitro over 48 hours exhibited increased hair follicle keratinisation when maintained in medium containing serum from rats aged 17 to 20 days, when compared to medium containing serum from rats aged 12 days of age.14 While follicles in rats aged 17 to 20 days would normally be in catagen/telogen, follicles from rats aged 12 days of age would be in anagen. This data suggests that serum from rats aged 17 to 20 days may contain some systemic hair follicle inhibitory factor. However, the specific nature of this inhibitory factor has not been determined although subsequent studies have shown that growth factors that may be present in serum, such as TGF-β, can inhibit hair follicle growth in vitro.16 Moreover, Paus et al. reported that the skin from telogen mice also contains an inhibitor of hair follicle growth although this also remains to be isolated.17
The reasons why hair follicle development and subsequent hair fibre production decreased with increasing age of embryo and why neonatal skin failed to sustain hair growth were not addressed in these early studies. However, a number of contributing factors were probably involved, including lack of appropriate tissue culture supplements, problems with diffusion, and possibly tissue damage on isolation. In recent years, significant advances have been made in the development and use of permeable substrates to support cultured skin. This and the increase in different types of culture medium and supplements available to the researcher have resulted in significant improvements in the quality of murine skin culture.18-23 However, long-term organ culture of human skin remains problematic as even 3mm punch biopsies are probably too large to allow diffusion of gases and nutrients, and as a result, cells within the biopsy undergo necrosis resulting in failure of the organ culture.
Traditionally, studies on individual isolated hair follicles were difficult due to the problems of obtaining sufficient numbers of follicles with which to work. Individual microdissected mouse hair follicles were used by Uzuka et al. to study increase in hair follicle bulb length with time in culture.24 Using time-lapse cinematography, these experiments showed that mouse hair follicles increased in length over 10 hours in culture. Collagenase digestion was used by Rogers et al. to isolate individual hair follicles from the skin of BALB/C mice.25 However, when the follicles were maintained in collagen matrices for 7 days, it was observed that they lost their morphology and formed cell aggregates, which may well have been a consequence of the disaggregation caused by the collagenase.
A significant advance in the field of hair follicle culture was made by Buhl et al. who showed that rat vibrissae follicles isolated by micro-dissection and maintained in vitro grew by 150μ over 3 days.26 Moreover, Buhl and his colleagues also showed that 0.5mM minoxidil stimulated whisker growth by up to 1mm over 3 days. These studies clearly demonstrated that organ maintenance of isolated, individual hair follicles was a viable alternative model to whole skin culture and that such models may be useful for investigating hair growth regulatory mechanisms. Following on from these studies, both our own group and others reported on the isolation of human hair follicles from facelift skin, and showed that these follicles could be successfully maintained in vitro.16,27
Hair Follicle Isolation
For the successful culture of hair follicles, it is essential to ensure that the hair follicles are not damaged on isolation. If follicles are even slightly damaged, then they will either fail to grow or more often grow at much reduced rates. A number of methods have been used to isolate intact hair follicles with variable results when the follicles are placed into organ culture. These include plucking, collagenase digestion, shearing, and micro-dissection.
Plucking. This is the oldest technique for isolating hair follicles and has been used as a source of ORS for primary and explant keratinocyte culture28-30 and for metabolic and cytotoxic experiments.31-33 However, as a source of hair follicles for organ maintenance, plucking has a number of drawbacks; namely, plucking usually results in considerable physical damage to the hair follicle and usually the dermal papilla and sebaceous gland are torn from the follicle and left in the skin.33-35 Because of this physical damage, plucked hair follicles are not viable for organ maintenance.
Collagenase digestion. Enzymes including collagenase have been used to isolate skin glands and appendages have been used by a number of workers.25,36-38 The major problem with any enzymatic form of isolation is that they can cause tissue disaggregation and promote primary cell outgrowth, neither of which are desirable in organ culture. Collagenase digestion, though, has been very useful for isolating large numbers of follicles for biochemical or cell culture.37,38
Shearing. Shearing was first reported for the successful isolation of human eccrine sweat glands39 and later for the isolation of human apocrine and sebaceous glands.40,41 The success of this method is due to the peri-glandular capsule of collagen that surrounds skin glands and appendages in situ. Repeated chopping of skin with a sharp pair of scissors causes this collagen capsule to shear and the glands to pop out. Shearing can be used to isolate intact human hair follicles,42 although the yield is variable. Because of the elongated nature of the hair follicle, as opposed to the globular structure of the skin glands, shearing tends to cut hair follicles into small segments, one of which usually includes an intact hair follicle bulb. Shearing has, however, proved very useful for the isolation of large numbers of intact, murine, pelage follicles for biochemical analysis.43
Micro-dissection. Traditionally, micro-dissection is the most reliable method of isolating intact hair follicles, although great care must be taken not to damage the follicle. At present, micro-dissection is the only accurate method for isolating intact pilosebaceous units consisting of the hair follicle and sebaceous gland. Sanders et al. have described the isolation of human pilosebaceous units from female facelift skin.44 It is obvious that while micro-dissection is a useful method for isolating intact pilosebaceous units, the number of follicles that can be obtained severely restricts the number of experiments that can be carried out. In 1990, we reported on a modified method of micro-dissection that was able to yield much larger numbers of intact hair follicle bulb.16 Isolation of hair follicles using this method is achieved by cutting human skin into thin strips approximately 3-5mm×10mm and then using a scalpel blade to cut through the skin at the dermal-subcutaneous fat interface. Then by placing the fat layer under a stereo dissecting microscope cut side uppermost, it is possible to identify the cut ends of the hair follicle protruding from the fat. Using watchmaker’s forceps, the outer root sheath can be grasped and the intact follicle bulb removed from the fat. Care must be taken when removing the follicles not to grasp the hair fibre as this will damage the follicle and dramatically influence the ability of these follicles to grow in culture.
Using this method, it is routinely possible to isolate in excess of 100 human anagen hair follicles in 1 to 2 hours from a piece of skin 4cm×2cm. Hair follicles in the catagen stage of their growth cycle are occasionally seen isolated by this method although their yield is small, usually 2 to 5 catagen follicles for every 100 anagen hair follicles isolated. These catagen hair follicles can be cultured and appear to progress into a telogen like state; however, their small number precludes any serious functional experiments. The method described above works very well for scalp hair follicles. Other workers have also reported isolation and in vitro culture of follicles from different areas of the body including the beard, axilla, and pubis, although no significant differences in growth rate were observed.45
The methods described above yield large numbers of hair follicle bulbs. However, by isolating follicles by cutting at the dermal-subcutaneous fat interface, we leave behind in the discarded epidermis and dermis most of the permanent portion of the hair follicle including the sebaceous gland, infundibulum, and isthmus. To isolate intact pilosebaceous units (PSUs), we use careful micro-dissection.44 These techniques are slow as care must be taken not to damage the hair follicle. However, by using such methods, it is possible to isolate several dozen PSUs in a couple of hours.
Human Hair Follicle Culture
Prior to the development of culture systems for human hair follicles, methods had been described for the maintenance of human sebaceous glands as well as apocrine and eccrine sweat glands using supplemented Williams E Medium,39 which was originally developed for the culture of hepatocytes.46 In our hands, Williams E Medium supports optimum growth of follicles when compared to other types of tissue culture medium, although a wide range of tissue culture medium and supplements have been used by researchers. Low calcium keratinocyte culture medium (KGM) does not support in vitro hair follicle growth, although the ORS remains viable. Histology of hair follicles cultured in KGM suggests that low-calcium-induced changes in cell adhesion are primarily responsible for the inhibition of hair growth. Likewise, culture of hair follicles in Williams E Medium lacking phenol red is also inhibitory, though whether this inhibition is a direct result of removing the estrogenic effects of phenol red is not known. Other media have been used by other workers including DMEM,27 Hams F12,47 RPMI16,40,48,49 and Eagles MEM.50 However, in all instances, lower rates of in vitro hair follicle growth were reported by these workers when compared to those reported for Williams E.
As well as selecting a suitable tissue culture medium for hair follicle culture, it is also essential to add additional supplements. In our hands, we have found that 2mM L-glutamine, Insulin (10μg/ml) and hydrocortisone (10-100ng/ml) are important for successful hair follicle culture. In addition, we also add penicillin (100U/ml) and streptomycin (100μg/ml). Insulin and glutamine are essential for successful in vitro follicle culture, and without their inclusion in tissue culture medium, very slow rates of follicle growth will be achieved. In addition, in the absence of insulin, some follicles will enter catagen.51 Penicillin and Streptomycin are our choice of antibiotics, although other workers have also used Gentamycin. Fungizone is inhibitory and should not be used. In our laboratories, we have found that serum inhibits hair follicle growth at levels as low as 1% (v/v) and have reported the optimum culture of isolated human hair follicles in defined serum free Williams E.52 However, other workers report stimulation of follicle growth by serum.27 This is difficult to interpret; however, it is worth noting that rates of in vitro growth achieved in these studies are lower than those achieved by us using serum free medium. Therefore, in suboptimal conditions, serum may indeed have a stimulatory action.
As well as a variety of different culture mediums and supplements having been used for follicle culture, a number of different support systems have also been used. In our laboratory, we traditionally place hair follicles in individual wells of 24 well plates. Imai et al. have used traditional methods of organ maintenance in which follicles were maintained at the air liquid interface on lens tissue paper placed on a stainless steel mesh.47,48 Kondo et al. used siliconized glass tubes containing 1.5ml of medium, gassed with 95% O2/5% CO2.27 Most tissue culture mediums are buffered by bicarbonate and therefore require gassing with between 5 and 10% CO2. In our hands, HEPES is inhibitory and to be avoided, if possible. Imai et al. have investigated a number of other different environments including 95% air/5% CO2 and 95% O2/5% CO2, and at different temperatures 25°C, 31°C, and 37oC of which they reported that 95% O2/5% CO2 and a temperature of 31°C were optimum.47 However, these observations were based on hair follicle morphology and rates of tritiated thymidine uptake, and hair follicle length measurements were not made. In our hands, gassing follicles with oxygen instead of air is toxic. Gelatin sponges have also been used to support human hair growth in vitro.50 We routinely use such sponges for maintenance of isolated PSUs44 and murine vibrissae follicles.53 However, we found that for isolated follicle bulbs, gelatin sponges do not offer any significant growth advantage.
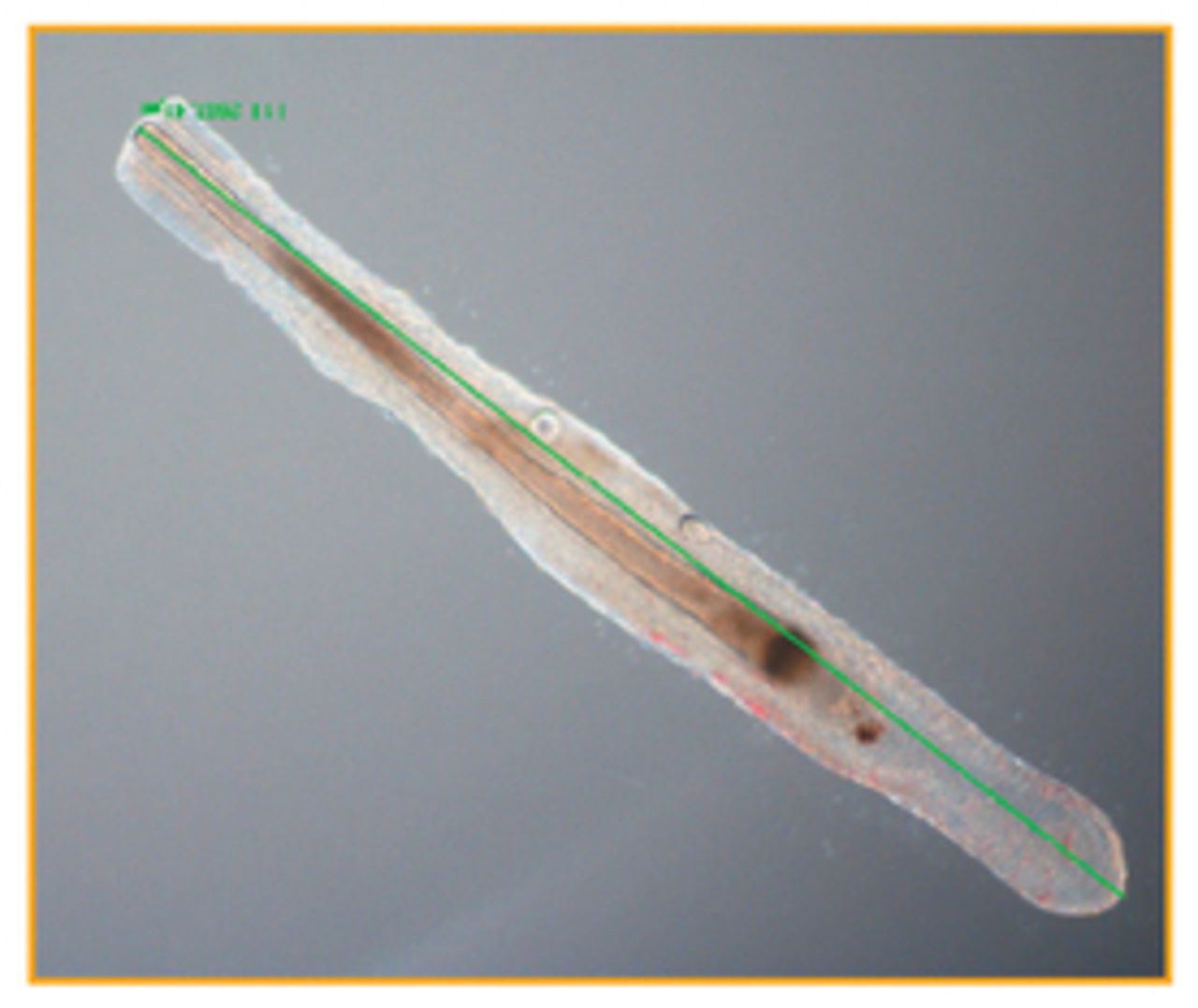
Most reports of hair follicle culture show that after 10 days follicle growth stops. This cessation is characterised by rounding of the DP, cessation of germinative epithelial proliferation, and keratinisation of the follicle bulb. In some papers, this has been referred to as a pseudo-catagen state (Figure 1). However, we would not recommend such terminology as these follicles show little similarity to a true catagen follicle as seen in vivo or in the absence of insulin in vitro (Figure 2). The reason why hair follicles stop growing in vitro after 10 days is not known. Cyclosporine A (CsA) has been reported to prolong follicle survival in vitro,54 and increasing hydrocortisone concentrations will also prolong follicle survival. However, even when using hydrocortisone or CsA, most follicles stop growing after 15 to 20 days. Individual follicles, however, have been cultured for up to 47 days, which suggests that prolonged culture may be possible especially with further development of tissue culture media and supplements and greater understanding of hair growth regulatory factors.


In a future Forum, I will review how hair follicle culture has been used to aid our understanding of hair biology and the future for such models.
- Copyright © 2013 by The International Society of Hair Restoration Surgery
References
In this issue




{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.